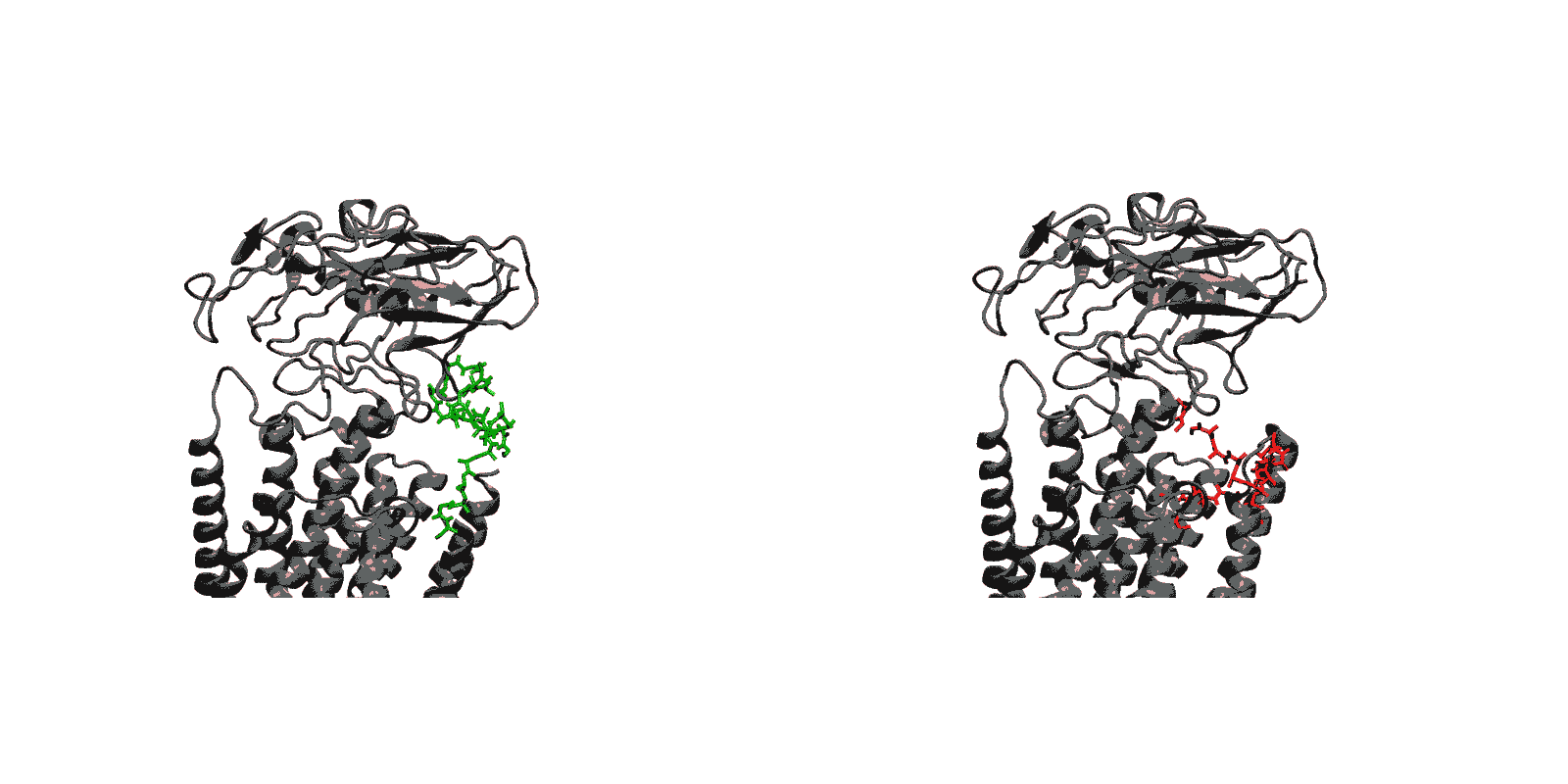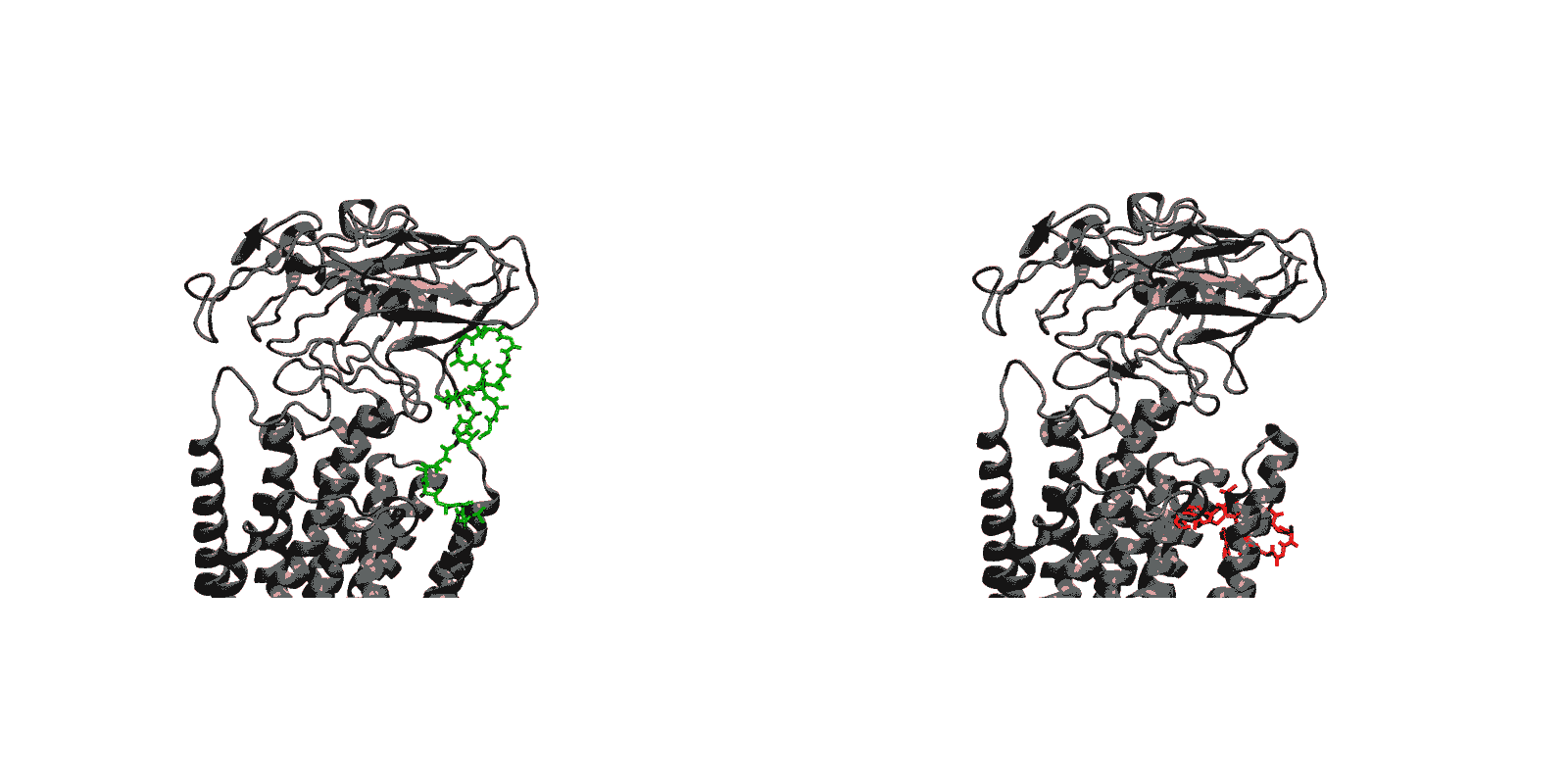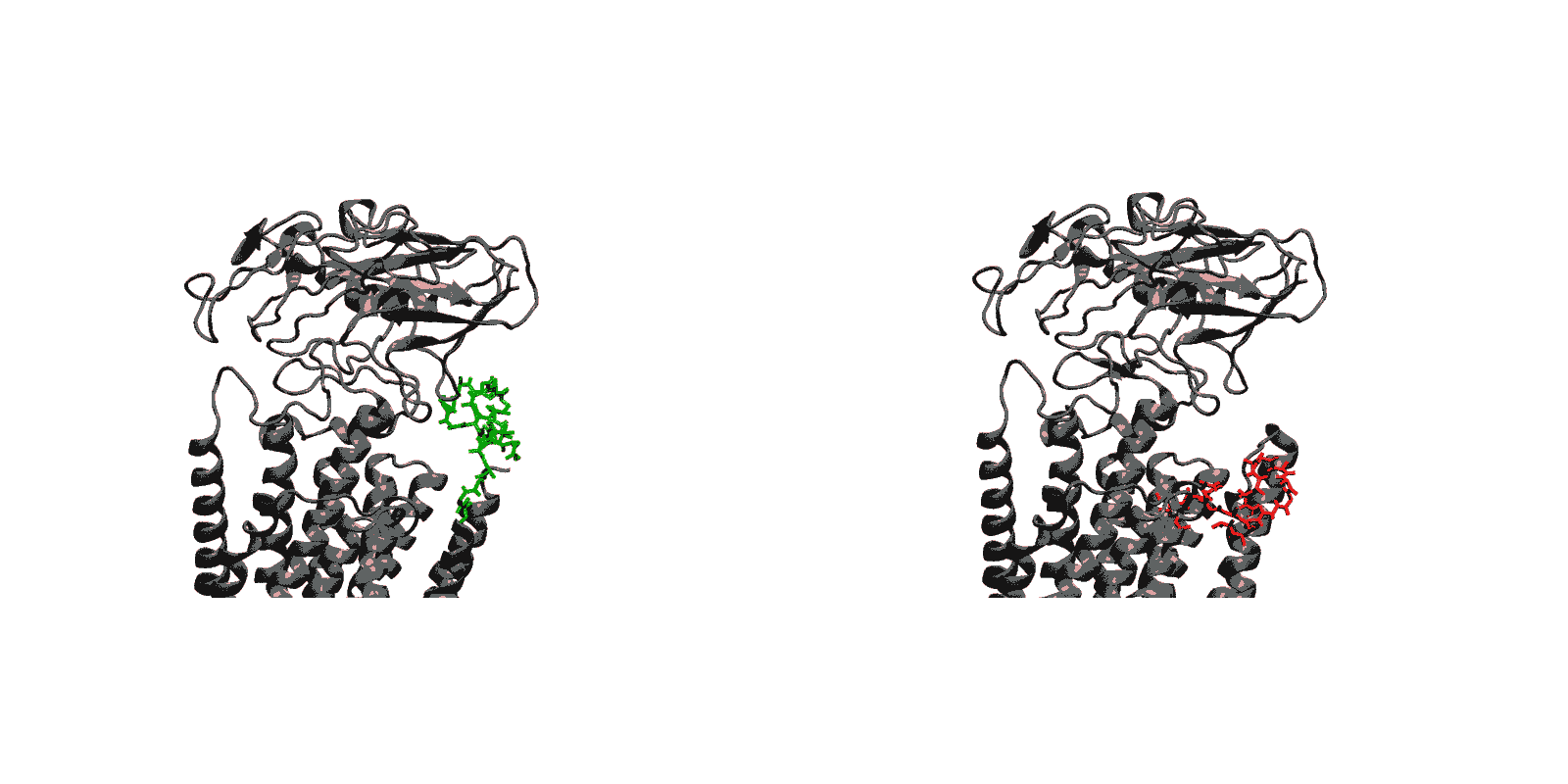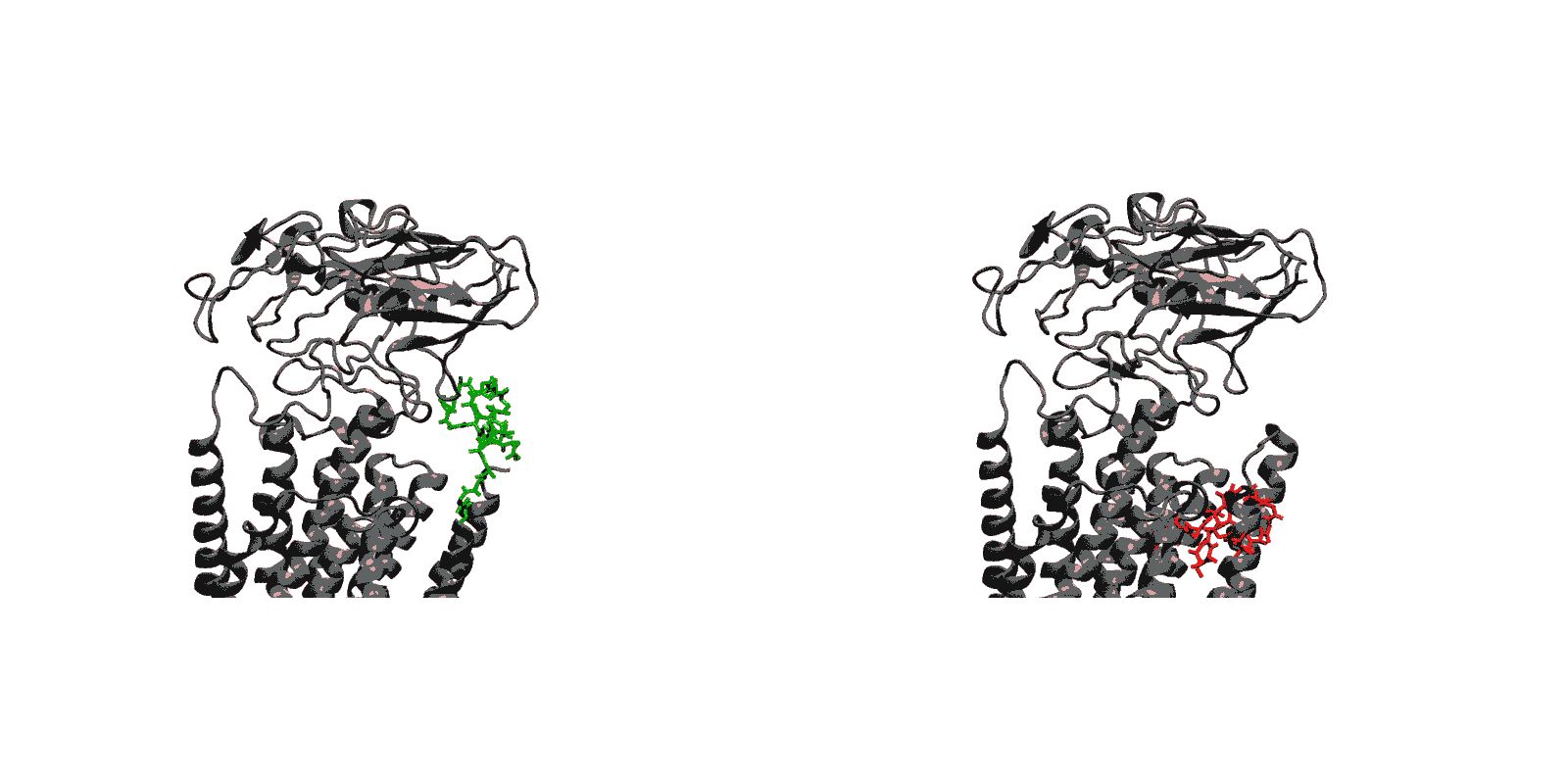Avisek Das, Huan Rui, Lei Huang, Robert Nakamoto & Benoît Roux
http://www.sciencedirect.com/science/article/pii/S0022283617300219
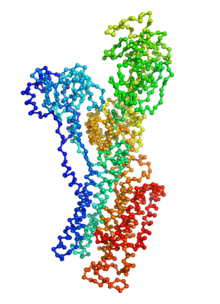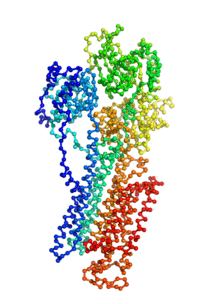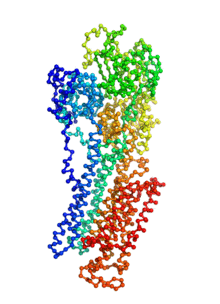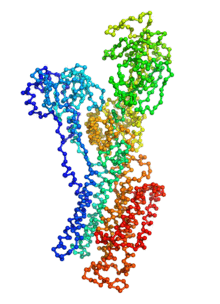
Ion pumps are integral membrane proteins responsible for transporting ions against concentration gradients across biological membranes. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), a member of the P-type ATPases family, transports two calcium ions per hydrolyzed ATP molecule via an “alternating-access” mechanism. While X-ray crystallography provides high-resolution snapshots about the stable experimentally resolved states of the transport cycle (Figure 1), it is very difficult to detect the short-lived intermediate conformations occurring transiently during the transport cycle.
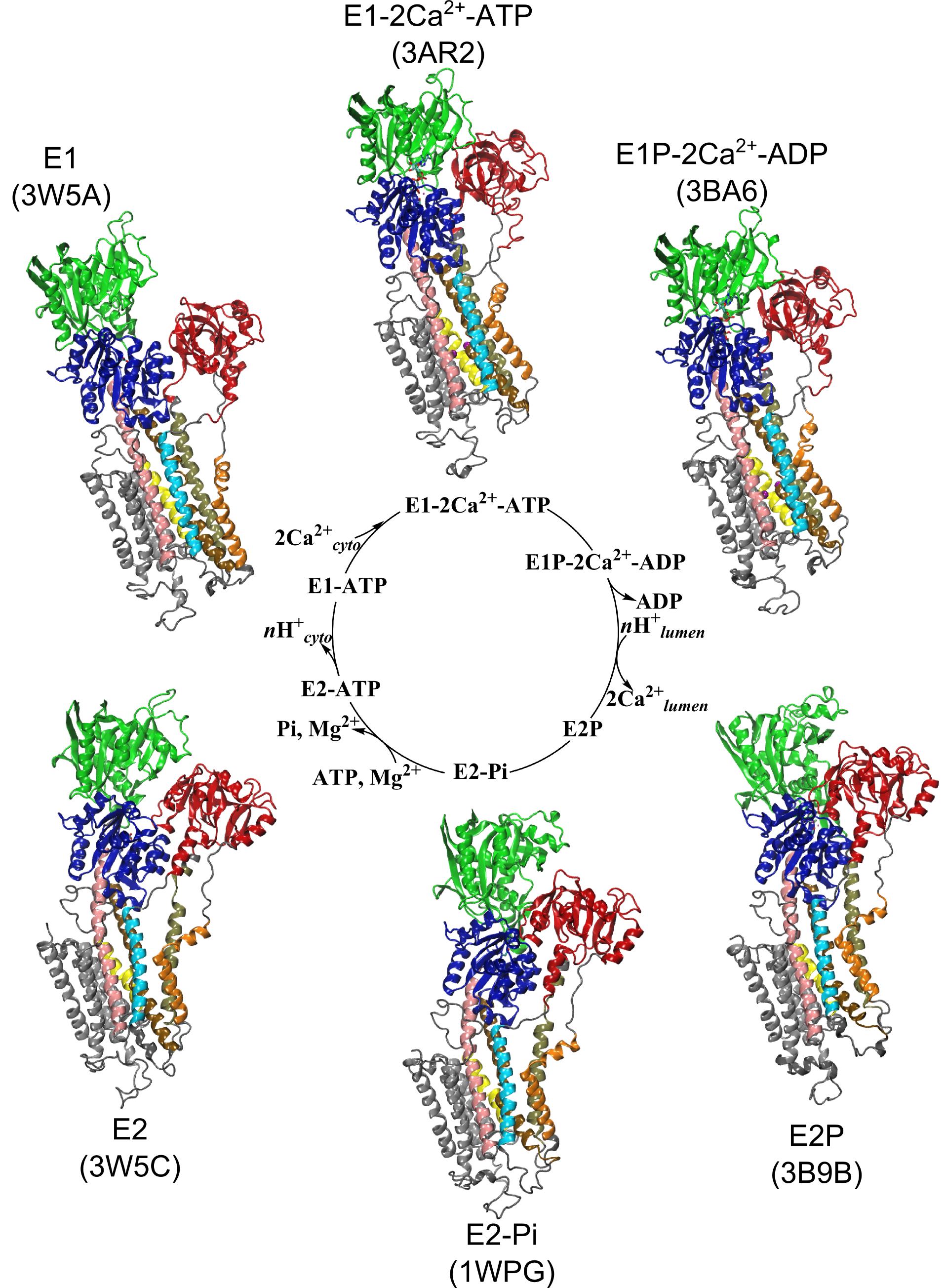
Figure 1: Structures of relevant functional states. The transport cycle of SERCA along with the structures of functional states used in the present work. The Protein Data Bank (PDB) id of each state is given in parenthesis below the state name.
Computational methods can be used to help supplement the missing information by providing atomic models for the transient intermediates along the transport cycle. Our goal with the present effort was to elucidate the details of the alternating access mechanism in SERCA by simulating the large-scale conformational transitions between the experimentally resolved stable sates. We calculated the pathway for three key steps:
- Cytoplasmic calcium binding and occlusion (E1 to E1-2Ca2+-ATP). Under normal physiological conditions, the pump is activated by the binding of Ca2+ ions and ATP, leading to the occlusion of the pump and the hydrolysis of ATP. A complete chronology of the binding of Ca2+ ions and the concomitant large-scale conformational changes leading to the occluded state are provided by the string pathway between the E1 and the E1-2Ca2+-ATP states.
- Luminal opening and calcium release (E1P-2Ca2+-ADP to E2P). Once the protein is stably locked in the occluded E1-2Ca2+-ATP state for an extended period of time, the phosphoryl transfer chemical reaction between ATP and Asp351 is expected to occur spontaneously, bringing the pump to the E1P-2Ca2+-ADP state (this reaction is not simulated here). This leads us to the next step in the transport cycle, which is the isomerization and conversion of the phosphorylated pump into the E2P state, where the luminal access channel is open.
- Closing of luminal gate and dephosphorylation (E2P to E2-Pi). After the release of Ca2+ ions, the next step in the transport cycle corresponds to the transition from the E2P state to the E2-Pi state, comprising two important molecular events: the closing of the luminal gate, and the dephosphorylation of Asp351 in the P domain.
Summary
The computational pathways provide a clear chronology of the key events underlying the active transport of calcium ions by SERCA. The main conclusions regarding three critical steps of the transport cycle E1→E1-2Ca-ATP→E2P→E2-Pi are:
- (E1 to E1-2Ca-ATP) Starting from the cytoplasmic open-access E1 state, the binding of ATP and the formation of the phosphoryl transfer catalytic site at the interface of the N and P domain together with the binding and sequestration of the two Ca2+ ions in the TM binding sites leading to occlusion are highly concerted and cooperative. The highly concerted transition explains the tight coupling between Ca2+ binding and ATP hydrolysis that is observed experimentally.
- (E1-2Ca-ATP to E2P) Phosphorylation of Asp351 when the pump is in the occluded E1-2Ca-ATP state drives the opening of the luminal gate via a two-step mechanism. In a first step, phosphorylation of Asp351 triggers a large-scale rotational movement of the cytoplasmic A domain to form a strong interaction with Asp351-P at the surface of the P domain (blocking of the catalytic site and preventing the back reaction by the ADP). In a second step, the large movement of the A domain is subsequently transmitted to the M4 helix via M3 helix leading to the disruption of the Ca2+ binding sites and the opening of the TM region toward the luminal side.
- (E2P to E2-Pi) The closing of the luminal gate following the dissociation of the Ca2+ ions to the luminal solution and protonation of binding sites residues is correlated with the dephosphorylation of Asp351-P via a two-step mechanism. In a first step, dehydration of the (protonated) binding sites together with the hydrophobic re-packing between the TM helices M4, M5 and M6 spontaneously drives the closing of the luminal gate. In a second step, the closed luminal gate mediated by the junction with the M2 helix induces a rotation in the position of the A domain, positioning the 181TGES loop in the A domain together with increased water access near Asp351-P needed for the dephosphorylation reaction.
Acknowledgments
This work was supported by the Membrane Protein Structural Dynamics Consortium funded by NIH/NIGMS through grant U54-GM087519. An award of computer time was provided by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program from the Department of Energy (DOE) of the United States of America. This research used resources of the Argonne Leadership Computing Facility (ALCF), which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357. Additional computational resources were provided by the Great Lake Consortium for Petascale Computing and the XSEDE program of the National Science Foundation of the United States of America.